Fluoreszenzmikroskopie

Die Fluoreszenzmikroskopie ist eine spezielle Form der Lichtmikroskopie. Sie beruht auf dem physikalischen Effekt der Fluoreszenz. Wenn fluoreszierende Stoffe mit Licht bestimmter Wellenlängen angeregt werden, strahlen sie Licht anderer, längerer Wellenlängen ab (Stokes-Verschiebung).
Bei der Fluoreszenzmikroskopie wird das erzeugte, vergrößerte Bild des untersuchten Objekts nur durch abgestrahltes (emittiertes) Licht erzeugt. Farbfilter verhindern, dass Anregungslicht auf das Bild gelangt. Fluoreszenzmikroskopische Bilder sind dann informativ, wenn nicht das ganze mikroskopische Präparat gleichmäßig fluoresziert, sondern wenn nur einige Strukturen leuchten. Diese Strukturen erzeugen helle Signale vor dunklem Hintergrund.
Jedes fluoreszierende Molekül im Präparat kann dabei als neue Lichtquelle angesehen werden. Liegt die Intensität der von diesen Molekülen abgestrahlten Fluoreszenz über der Nachweisgrenze, können mit der Fluoreszenzmikroskopie auch Strukturen nachgewiesen werden, die weit kleiner sind als die Auflösungsgrenze des Mikroskops. Die Auflösungsgrenze wird bei der klassischen Fluoreszenzmikroskopie aber nicht überwunden, da bei kleinem Abstand zwar ein Nachweis möglich ist, aber keine Aussage darüber, ob das Signal von einer oder von mehreren Strukturen hervorgerufen wird. In dieser Hinsicht besteht Ähnlichkeit zur Dunkelfeldmikroskopie.
Neben der klassischen gibt es zahlreiche weiterentwickelte Spezialformen der Fluoreszenzmikroskopie. Hierzu gehören beispielsweise die konfokale Laserscanningmikroskopie und die Multi-Photonen-Fluoreszenzmikroskopie. Ab den 1990er-Jahren wurden verschiedene Verfahren entwickelt, die tatsächlich eine deutlich verbesserte Auflösung ermöglichen. Diese sogenannten Höchstauflösungs- oder Superresolution-Verfahren sind ebenfalls fluoreszenzmikroskopischer Art.[1]
Grundlagen
Fluoreszenz


Ein fluoreszierendes Molekül hat ein Elektron, das durch Absorption eines Photons von einem energiearmen Grundzustand (S0) in einen energiereicheren, angeregten Zustand (S1) übergehen kann. Sowohl S0 als auch S1 haben mehrere Unterzustände, die sich jeweils im Gehalt der Schwingungsenergie (auch: Vibrationsenergie) des Elektrons unterscheiden. Der Energieunterschied zwischen dem Ausgangs-Schwingungszustand innerhalb von S0 und dem erreichten Schwingungszustand in S1 entspricht genau dem Energiegehalt des absorbierten Photons.
Fällt das Elektron auf einen Grundzustand zurück, wird ein Photon ausgesendet. Diese Lichtemission erfolgt wenige Nanosekunden nach der Absorption – das ist die Fluoreszenz. Damit sie zustande kommt, muss zwischen S0 und S1 ein deutlicher Unterschied im Energiegehalt liegen und es dürfen keine weiteren Energieniveaus dazwischen liegen, da angeregte Elektronen sonst über nichtstrahlende Prozesse in den Grundzustand zurückkehren.[2]
Da sowohl der Grundzustand S0 als auch der angeregte Zustand S1 mehrere Unterzustände haben, können nicht nur Photonen mit genau einem bestimmten Energiegehalt absorbiert oder emittiert werden, sondern auch Photonen mit ähnlichen Energiegehalten. Da sich der Energiegehalt eines Photons umgekehrt proportional zu seiner Wellenlänge verhält, bedeutet das, dass ein fluoreszierender Stoff durch einige ähnliche Wellenlängen angeregt wird: Man spricht vom Anregungsspektrum. Genauso strahlt er einige ähnliche Wellenlängen ab, das Emissionsspektrum.[2]
Das Abstrahlen der Fluoreszenz geschieht grundsätzlich vom niedrigsten angeregten Energieniveau aus (Kasha-Regel). Wird das Elektron durch die Absorption des Anregungsphotons auf einen höheren angeregten Zustand gehoben, so gelangt es zunächst durch nichtstrahlende Energieabgabe auf das niedrigste angeregte Energieniveau, bevor es zur Emission eines Photons kommt. Dies hat für die Fluoreszenzmikroskopie mehrere wichtige Konsequenzen:[2]
|
Stoffe, die fluoreszieren, werden als Fluorophore bezeichnet. Fluorophore, die verwendet werden um Präparate anzufärben, werden als Fluoreszenzfarbstoffe oder Fluorochrome bezeichnet.
Autofluoreszenz und Fluorochrome


Wenn ein Präparat von selbst fluoresziert, wird dies als Autofluoreszenz, Eigenfluoreszenz oder Primärfluoreszenz bezeichnet. Viele Pflanzen haben in verschiedenen Teilen sehr starke Autofluoreszenz, zum Beispiel Samenpflanzen in den hölzernen Teilen ihrer Sprossachsen. Das Chlorophyll in den Chloroplasten der grünen Pflanzenzellen ist stark rot fluoreszierend. Tierische Zellen fluoreszieren im Vergleich dazu nur schwach, jedoch noch stark genug, um Fluoreszenzmarkierungen unter Umständen zu verschleiern. Die Hauptquellen hier sind Flavine, die in den Mitochondrien vorkommen und Lipofuscin in den Lysosomen.[3] Das Coenzym NADPH zeigt ebenfalls Autofluoreszenz.[4]
Eine in einem Präparat mit Fluorochromen künstlich erzeugte Fluoreszenz ist eine Sekundärfluoreszenz. Der Prozess, der dazu führt, heißt Fluoreszenzmarkierung. Gute Fluorochrome vereinigen mehrere Eigenschaften: (1) Sie haben eine hohe Wahrscheinlichkeit ein Photon zu absorbieren, das heißt, sie haben einen hohen Absorptionskoeffizienten. (2) Die meisten der absorbierten Photonen führen tatsächlich zur Emission eines Flureszenzphotons (hohe Quanteneffizienz). Beides zusammen führt zu einer großen Helligkeit. (3) Fluorochrome sollten ein geringes Bleichen aufweisen, das heißt, dass sie sich oft anregen lassen, ohne zerstört zu werden. (4) Außerdem sollten Fluorochrome in einem möglichst schmalen Bereich des Lichtspektrums fluoreszieren, damit möglichst viele Fluorochrome mit unterschiedlichen Fluoreszenzfarben gleichzeitig verwendet werden können, um unterschiedliche Strukturen anzufärben.[2]
Farbkanäle

Wenn sich die Anregungs- und Emissionsspektren zweier Fluorochrome stark überlappen, dann können diese nicht voneinander unterschieden werden. Beispielsweise haben Fluorescein, Grün fluoreszierendes Protein, Spectrum Green und eine Reihe weiterer kommerziell erhältlicher Farbstoffe sehr ähnliche Spektren, so dass sie anhand ihrer Fluoreszenzfarbe nicht unterschieden werden können. Wenn verschiedene Fluoreszenzfarbstoffe nebeneinander eingesetzt werden sollen, um verschiedene Strukturen anzufärben, müssen diese Farbstoffe unterschiedliche Spektren haben. Typischerweise regt UV-Licht blau fluoreszierende Fluorochrome an, blaues Licht grüne Fluorochrome und grünes Licht rote Fluorochrome. Diese drei Farbkanäle lassen sich also verwenden, um im gleichen Präparat unterschiedliche Strukturen darzustellen.
Damit ist die Zahl der gleichzeitig nachweisbaren Farben jedoch nicht erschöpft. Acht verschiedene Fluorochrome wurden bereits parallel eingesetzt. Dazu verwendete man DAPI als blau fluoreszierende Gegenfärbung für DNA sowie sieben weitere Farbstoffe, die an Gensonden für Fluoreszenz-in-situ-Hybridisierung gekoppelt wurden: Diethylaminocoumarin (Deac), Spectrum Green und die Cyanin-Farbstoffe Cy3, Cy3.5, Cy5, Cy5.5 und Cy7.[5][6] Die meisten Fluoreszenzmikroskope haben drei bis fünf Farbkanäle.
Aufbau von Fluoreszenzmikroskopen
Bei der „normalen“ Lichtmikroskopie, der Durchlicht-Hellfeldmikroskopie, wird das Bild durch Licht erzeugt, welches das Präparat durchstrahlt. Dies ist bei der Fluoreszenzmikroskopie nicht der Fall. Hier wird das Bild durch Fluoreszenzlicht erzeugt, das erst im Präparat entsteht. Das Anregungslicht, welches das Präparat bestrahlt, wird dagegen durch spezielle Filter von der Bilderzeugung ausgeschlossen. Da sich Fluoreszenzlicht unter normalen Bedingungen gleichmäßig in alle Raumrichtungen ausbreitet, ist es daher grundsätzlich egal, ob das Anregungslicht von oben, von unten oder von der Seite kommt. Tatsächlich wurden alle drei Varianten umgesetzt.
Zu Beginn der Fluoreszenzmikroskopie, in der ersten Hälfte des 20. Jahrhunderts, wurden Durchlicht-Fluoreszenzmikroskope gebaut. Sie sind heute nur noch von historischem Interesse (siehe Abschnitt Geschichte unten). Durch die Verfügbarkeit Dichroitischer Spiegel (auch: dichroitischer Strahlteiler) wurde es ab etwa 1970 möglich, Auflicht-Fluoreszenzmikroskope zu bauen, bei denen das Anregungslicht über das Objektiv in das Präparat eingestrahlt wird. Sie werden auch Epifluoreszenzmikroskope genannt, nach griechisch ἐπί für „auf“. Seit Ende des 20. Jahrhunderts wird dieser Bautyp fast ausschließlich verwendet. Die seitliche Beleuchtung findet bei einem spezialisierten Fluoreszenzmikroskop Anwendung, dem Lichtscheibenmikroskop (siehe unten).
Das Epifluoreszenzmikroskop: das typische Fluoreszenzmikroskop
Im Vergleich zu Durchlicht-Hellfeldmikroskopen haben Epifluoreszenzmikroskope eine zusätzliche Auflicht-Beleuchtungsachse für das Fluoreszenz-Anregungslicht. Das zu beobachtende Objekt wird durch das Objektiv beleuchtet. Die folgende Nummerierung bezieht sich auf die Schemazeichnung.
- Durch einen passend ausgewählten optischen Filter, den Anregungsfilter, wird von der verwendeten Lampe nur der Teil des erzeugten Lichts durchgelassen, der die für die Anregung des Fluoreszenzfarbstoffs notwendigen Wellenlängen enthält. Der Bereich des Spektrums, in welchem der Fluoreszenzfarbstoff leuchtet, darf vom Anregungsfilter nicht durchgelassen werden.
- Ein Strahlteiler spiegelt das Anregungslicht zum Objektiv, worauf im Präparat die Fluoreszenz entsteht. Das Objektiv übernimmt also auch die Funktion des Kondensors. Die Fluoreszenz ist langwelliger als das Anregungslicht.
- Der Anteil des Fluoreszenzlichts, der vom Objektiv gesammelt wird, gelangt wiederum zum Strahlteiler. Durch dessen besondere Eigenschaften wird dieses längerwellige Licht in Richtung Okular oder Detektor durchgelassen (und nicht gespiegelt). Anregungslicht, das im Präparat reflektiert wird, wird dagegen weitgehend wieder zur Lampe gelenkt.
- Da Strahlteiler nicht ganz perfekt arbeiten, gelangt ein geringer Teil des im Präparat reflektierten Anregungslichts trotzdem in Richtung Okular beziehungsweise Detektor. Da die Intensität der Fluoreszenz im Vergleich zur Anregung sehr schwach ist, ist deshalb ein weiterer optischer Filter, genannt Sperrfilter oder Emissionsfilter, erforderlich, um dieses restliche Anregungslicht zu eliminieren.[1][2]
Die drei genannten Filter sind in heutigen Fluoreszenzmikroskopen oft in einen gemeinsamen Block eingebaut. Dieser befindet sich bei aufrechten Mikroskopen in der optischen Achse über dem Objektiv. Bei inversen Fluoreszenzmikroskopen befindet er sich entsprechend unter dem Objektiv. Bei Geräten mit Unendlich-Optik liegt er im Unendlichraum zwischen Objektiv und Tubuslinse.
 Schema eines Epifluoreszenzmikroskops
Schema eines Epifluoreszenzmikroskops Ein aufrechtes Epifluoreszenzmikroskop. Das schwarze Gehäuse rechts enthält die Lichtquelle für die Fluoreszenz-Anregung. Im schwarzen Gehäuse oben auf dem Mikroskop befindet sich eine CCD-Kamera.
Ein aufrechtes Epifluoreszenzmikroskop. Das schwarze Gehäuse rechts enthält die Lichtquelle für die Fluoreszenz-Anregung. Im schwarzen Gehäuse oben auf dem Mikroskop befindet sich eine CCD-Kamera. Aufrechtes Epifluoreszenzmikroskop mit offener Abdeckung und Blick auf den Revolver mit Fluoreszenzfilter-Würfeln
Aufrechtes Epifluoreszenzmikroskop mit offener Abdeckung und Blick auf den Revolver mit Fluoreszenzfilter-Würfeln Inverses Epifluoreszenzmikroskop. Das Objektiv befindet sich unter der Präparatposition und ist hier nicht sichtbar.
Inverses Epifluoreszenzmikroskop. Das Objektiv befindet sich unter der Präparatposition und ist hier nicht sichtbar.
Lichtquellen
Die Erzeugung der Fluoreszenz im Präparat ist kein effektiver Prozess: Nur ein Bruchteil des Anregungslichts wird von den Fluoreszenzfarbstoffen absorbiert. Um trotzdem helle, mit dem Auge sichtbare Signale erzeugen zu können, sind daher sehr hohe Leuchtstärken erforderlich.[2]
Typischerweise sind Fluoreszenzmikroskope mit Quecksilberdampflampen, Halogenmetalldampflampen, Xenon-Gasentladungslampen oder, seit dem 21. Jahrhundert, mit LED-Lampen ausgestattet. Die meisten Lichtquellen leuchten über das gesamte sichtbare Spektrum sowie im ultravioletten Bereich. Die für das jeweils zu untersuchende Fluorochrom erforderlichen Wellenlängen werden durch einen entsprechenden Filter ausgewählt und alle anderen unterdrückt.[2]
Filter


Während früher Farbfilter aus gefärbtem Glas zum Einsatz kamen, werden heute oft Interferenzfilter verwendet. Interferenzfilter sind jedoch deutlich teurer, so dass gefärbtes Glas immer noch zur Anwendung kommt. Für Anregungs- und Emissionsfilter können beide Typen verwendet werden. Der dichroitische Strahlteiler kann nur als Interferenzfilter hergestellt werden.[2]
Interferenzfilter bestehen aus einer Glasscheibe, auf die mehrere dünne Materialschichten aufgetragen werden. Zwischen den Schichten entstehen Interferenzen, so dass bestimmte Wellenlängen durchgelassen werden, andere aber gespiegelt werden. Im Gegensatz zu farbigem Glas wird das Licht also nicht absorbiert. Durch die Wahl geeigneter Materialien und Schichtdicken können Filter für unterschiedliche Wellenlängen hergestellt werden. Licht, das in unterschiedlichen Winkeln auf Interferenzfilter auftrifft, legt unterschiedlich lange Strecken in den jeweiligen Schichten zurück. Daher ändern sich die Filtereigenschaften in Abhängigkeit vom Einfallswinkel des Lichts. Ein Interferenzfilter muss deswegen im vorgesehenen Winkel im Mikroskop eingebaut werden, um richtig zu funktionieren.[7]
Von der Funktionsweise her werden Kurzpass-, Langpass- und Bandpass-Filter unterschieden. Kurzpassfilter lassen Licht bis zu einer bestimmten Wellenlänge durch. Ein KP480 würde also Licht bis zu 480 nm durchlassen und Licht längerer Wellenlängen blockieren. Im Gegensatz dazu lassen Langpass-Filter Licht ab einer bestimmten Wellenlänge durch, ein LP520 also Licht mit Wellenlängen länger als 520 nm. Bandpass-Filter lassen nur einen bestimmten Abschnitt des Spektrums durch. Ein Bandpassfilter mit der Bezeichnung 525/50 lässt ein spektrales Fenster von 50 nm passieren, dessen Mitte bei 525 nm liegt, also ein Fenster von 500–550 nm. Die Eigenschaften eines Filters werden meist nur auf den sichtbaren Bereich und direkt angrenzende spektrale Bereiche bezogen. Es kann daher sein, dass zum Beispiel ein Bandpassfilter für den sichtbaren Bereich im Infrarot wieder durchlässig wird. Dies kann bei Zwei-Photonen-Fluoreszenzanregung (siehe unten) zu Problemen führen.[7]
Mit Interferenzfiltern lassen sich auch komplexere spektrale Eigenschaften realisieren. Beispielsweise kann ein Filter mehrere spektrale Fenster durchlassen, die Bereiche dazwischen aber blockieren (Multi-Bandpass). Auch dichroitische Strahlteiler, die mehrere spektrale Bereiche spiegeln und dazwischen liegende Bereiche durchlassen, sind machbar (Multi-Dichroic). Dadurch wird es möglich, mehrere Fluoreszenzkanäle gleichzeitig zu sehen. Manche Lichtquellen können sehr schnell zwischen verschiedenen Anregungswellenlängen hin- und herschalten, zum Beispiel einige LED-Geräte. Unter Verwendung eines Multi-Dichroics und eines Multi-Bandpassfilters als Emissionsfilter können verschiedene Fluoreszenzkanäle sehr schnell abwechselnd verwendet werden, da keine Filter bewegt werden müssen.[2]
In Epifluoreszenzmikroskopen sind Anregungsfilter, Strahlteiler und Emissionsfilter meist zu Filterblöcken zusammengefasst. Von einem Kanal zum anderen werden dabei alle drei Filter gemeinsam ausgetauscht, indem man entweder einen Schieber verschiebt, in dem Kombinationen für drei bis vier Farben enthalten sind, oder indem ein Rad gedreht wird, auf dem mehrere Filterwürfel für jeweils einen Kanal montiert sind.
In manchen Laserscanningmikroskopen, die mit Fluoreszenz funktionieren, wird die Funktion von Anregungsfiltern oder Strahlteilern durch Akustooptische Modulatoren (AOM, auch: acousto-optical tunable filter (AOTF) oder acousto-optical beam splitter (AOBS)) ersetzt. Der Emissionsfilter kann ersetzt werden, indem das Fluoreszenzlicht, das in diesen Geräten nur von einem Punkt kommt, vor der Detektion spektral aufgetrennt wird und dann nur gewünschte Teile des Spektrums detektiert werden. Eine solche Auftrennung wird mit einem Prisma oder einem Beugungsgitter erreicht.[8]
Detektoren
Präparate mit heller Fluoreszenz im für das menschliche Auge gut sichtbaren Bereich bis etwa 620 nm Wellenlänge können direkt durch das Okular betrachtet werden. Zur Dokumentation werden Kameras eingesetzt. Während früher fotografischer Film verwendet wurde, fand Ende des 20. Jahrhunderts eine Umstellung auf elektronische Kameras statt. Häufig werden CCD-Kameras verwendet, die Schwarzweißbilder aufnehmen. Durch den Verzicht auf Farbfilter in der Kamera können alle Pixel jede Farbe aufnehmen, das Bild wird heller, als es bei Farbkameras der Fall wäre. Welche Farbe von der Kamera tatsächlich aufgenommen wird, wird durch den vorgeschalteten Emissionsfilter festgelegt. Verschiedenfarbige Fluoreszenzfarbstoffe in einem Präparat werden nacheinander aufgenommen und können im Computer übereinander gelegt werden. Dabei kann den jeweiligen Farbstoffen eine beliebige Farbe zugeordnet werden, wahlweise ihre natürliche Farbe oder eine andere, letzteres etwa um Farben besser zu kontrastieren.[9][10]
Schwierigkeiten bei der Fluoreszenzmikroskopie
Ausbleichen der Fluoreszenz


Fluoreszierende Moleküle lassen sich nicht beliebig oft anregen, da sie durch das Anregungslicht zerstört werden können. Der als Photobleichung bezeichnete Prozess geschieht je nach Photostabilität der fluoreszierenden Präparate schneller oder langsamer. Für ein einzelnes fluoreszierendes Molekül ist Ausbleichen dann mehr oder weniger wahrscheinlich.
Ein Molekül im angeregten Zustand kann nicht nur durch Fluoreszenz wieder in den Grundzustand (S0) übergehen. Eine zweite Möglichkeit ist die Innere Umwandlung, bei der die Energie beim Übergang in den Grundzustand in Wärme umgewandelt wird. Eine dritte ist das Ausbleichen, bei der es zur Zerstörung des Farbstoffs durch photochemische Reaktionen kommt. Diese Reaktionen hängen mit den Spin-Zuständen der Elektronen zusammen.
Die Elektronen eines Fluorochroms befinden sich normalerweise in einem Singulett-Zustand, in dem alle Elektronen des Moleküls Spin-gepaart sind. Daher wird der Grundzustand auch als S0, der „normale“ angeregte Zustand als S1 bezeichnet (siehe Abbildung mit Energiediagramm). Wie oben beschrieben haben S0 und S1 mehrere Unterzustände, die sich im Energieniveau jeweils wenig unterscheiden. Zusätzlich gibt es aber weitere, noch energiereichere angeregte Zustände, die im Energieniveau deutlich höher liegen als S1. Sie werden als S2, S3 und so weiter bezeichnet. Auch diese haben mehrere Unterzustände. Durch ein energiereiches Photon kann ein Molekül aus dem Grundzustand auch in einen dieser Zustände befördert werden. Das Absorptionsspektrum zeigt dann bei Wellenlängen mit dem entsprechenden Energiegehalt ein lokales Maximum (siehe Abbildung der Spektren). Das Fluoreszenzspektrum bleibt aber gleich, da das Fluoreszenz-Photon immer vom niedrigsten angeregten Zustand ausgesandt wird (siehe auch oben).[2]
Neben den Singulett-Zuständen kommen auch Triplett-Zustände vor. Der energieärmste Triplett-Zustand wird als T1 bezeichnet (siehe Energiediagramm), energiereichere als T2, T3 und so weiter. Der Übergang von einem Singulett- in einen Triplett-Zustand wird als Intersystem Crossing bezeichnet. Bei diesem Übergang muss sich der Spin eines Elektrons umdrehen, so dass dann ein ungepaartes Set von Elektronenspins vorliegt. Die Wahrscheinlichkeit hierfür ist normalerweise gering, steigt aber deutlich, wenn sich das Molekül in einem der höheren angeregten Zustände befindet, also S2 oder höher. Um die Wahrscheinlichkeit für einen Übergang zu den Triplett-Zuständen zu minimieren, sollte die Anregung nach S2 oder höher nach Möglichkeit vermieden werden, denn aus Triplett-Zuständen kann keine Fluoreszenz entstehen, und diese Zustände sind langlebig.[2]
Aus dem Triplett-Zustand kann das Elektron seine Energie entweder als Wärme abgeben, um wieder in den Grundzustand zu gelangen, oder es gibt ein Photon ab. Dieses Photon entsteht deutlich später nach der Anregung als Fluoreszenz, es handelt sich um Phosphoreszenz.
Aus dem Triplett-Zustand heraus kann das Fluorochrom aber auch zerstört werden, also Ausbleichen. Im Gegensatz zu den angeregten Singulett-Zuständen ist die Verweildauer im Triplett-Zustand deutlich länger, daher ergibt sich hier eher die Möglichkeit mit anderen Molekülen der Umgebung chemisch zu reagieren. Hat der Fluoreszenzfarbstoff chemisch reagiert, ist das Reaktionsprodukt in der Regel nicht fluoreszierend und der Farbstoff ist ausgebleicht. Von besonderer Bedeutung ist die Reaktion mit molekularem Sauerstoff, O2, da dieser einen Triplett-Grundzustand hat und Triplett-Triplett-Reaktionen sehr effektiv ablaufen.[2]
Bleichen kann daher verringert werden, wenn Sauerstoff aus dem Präparat entfernt wird. Dies kann durch sogenannte Antifade-Substanzen (englisch für Antibleichmittel) erreicht werden, die reduzierende Wirkung haben. Zum Einsatz kommen Antioxidantien, zum Beispiel para-Phenylamin-Diamin, DABCO oder Gallate.[2] Ganz lässt sich das Bleichen jedoch nicht verhindern. Daher ist es wichtig, für die Fluoreszenzmarkierung Fluorochrome auszuwählen, die möglichst photostabil sind.[11]
Phototoxizität
Die im vorigen Abschnitt beschriebenen Reaktionen treten auch bei der Fluoreszenzmikroskopie von lebenden Zellen oder Organen auf. Hier kommt es nicht nur zum Ausbleichen, sondern die bei der Reaktion mit Sauerstoff entstehenden reaktiven Sauerstoffspezies können in einer Nachfolgereaktion zelluläre Komponenten schädigen und so zum Tod der Zelle führen. Auch hier kann die Zugabe von reduzierenden Stoffen helfen, etwa Ascorbinsäure oder Trolox.[12][13]
Kurzwelliges Licht, besonders UV-Licht, kann Zellen aber auch direkt schädigen, ohne dass Fluoreszenz hervor gerufen wird. Dies stellt ein Problem bei Lichtquellen mit breitem Spektrum und hohem UV-Anteil dar, wie bei Quecksilberdampflampen. Der hohe UV-Anteil der Lichtquelle kann durch Filter nicht vollständig blockiert werden. Die Verwendung von UV-freien Lichtquellen wie LED, Lasern oder Halogenlampen ist daher für Lebendbeobachtungen von Vorteil.
Beide Probleme verringern sich, wenn die Belichtung für die jeweilige Fragestellung so gering wie möglich gehalten wird. Hierzu können auch besonders empfindliche Kameras oder andere Detektoren beitragen. Die Bildung reaktiver Sauerstoffspezies kann verringert werden, wenn das Aufsuchen des Bildbereichs (Zellen) und das Fokussieren nicht mit Fluoreszenz, sondern mit Hellfeld, Phasenkontrast, Differentialinterferenzkontrast oder vergleichbaren Verfahren erfolgt und die fluoreszenzmikroskopische Untersuchung dadurch auf ein Minimum beschränkt wird.[12][13]
Neben den genannten phototoxischen Effekten kann es bei zu hohen Konzentrationen der für die Fluoreszenzmarkierung verwendeten Substanzen zur direkten Vergiftung der Zellen kommen.[13]
Übersprechen von Signalen in benachbarte Farbkanäle
Als bleedthrough (englisch für durchbluten) oder crosstalk, also Übersprechen, wird es bezeichnet, wenn bei einer Mehrfachmarkierung ein Signal auch im benachbarten Farbkanal zu sehen ist. Das langwellige Ende des Emissionsspektrums der meisten Fluorochrome fällt nur sehr langsam gegen Null ab. Daher kommt es bei gleichzeitiger Anregung und Detektion von im Spektrum benachbarten Fluorochromen häufig zum Übersprechen, beispielsweise eines grünen Fluorochroms in den benachbarten orangefarbenen Kanal. Dies kann verhindert oder zumindest vermindert werden, wenn enge Bandpass-Filter verwendet werden und/oder die Farbstoffe nicht gleichzeitig, sondern nacheinander angeregt werden.[14]
Präparateerstellung und Anwendungen in den Lebenswissenschaften
In den Biowissenschaften wird Fluoreszenzmikroskopie vielfältig eingesetzt. Manche zu untersuchende Objekte sind von selbst fluoreszierend. Dies wird als Autofluoreszenz bezeichnet. Beispielsweise haben Pflanzen Chlorophylle und andere Pigmente, die natürlicherweise fluoreszieren. Autofluoreszenz ist aber häufig unerwünscht, da sie als Hintergrundfluoreszenz das Erkennen von künstlichen Fluoreszenzen erschwert.
Für biomedizinische Anwendungen ist eine Vielzahl von Methoden für die Fluoreszenzmarkierung entwickelt worden. Manche Fluoreszenzfarbstoffe binden auf Grund ihrer chemischen Eigenschaften direkt an die zu untersuchende Struktur. In diese Gruppe fallen beispielsweise DNA-Farbstoffe wie DAPI, Acridinorange und Hoechst 33342 oder Membranfarbstoffe wie Nilrot oder DiI. Einige solcher Farbstoffe können auch lebende Zellen anfärben.[15]
In anderen Fällen wird ein nicht-fluoreszierendes Molekül, das an die zu untersuchende Struktur bindet, chemisch mit einem Fluoreszenzfarbstoff gekoppelt. Beispiele für diesen Ansatz sind die Immunfluoreszenz, bei der Fluoreszenz-markierte Antikörper verwendet werden, die Fluoreszenz-in-situ-Hybridisierung, eine Technik zum mikroskopischen Nachweis von größeren DNA-Abschnitten, oder die Färbung von Aktin mit fluoreszenzmarkiertem Phalloidin.[15]
Proteine können gentechnisch mit einem fluoreszierenden Protein wie GFP fusioniert werden. Aus dem fluoreszenzmikroskopischen Bild können anschließend Rückschlüsse auf die Verteilung und Anordnung des untersuchten Proteins in der lebenden und meist fixierten Zelle gezogen werden, (etwa im Zellkern, im Cytoplasma, Zellmembran-gebunden oder nach außen exportiert) beziehungsweise Zellbestandteile durch ihre spezifischen Proteine visualisiert werden (wie Aktinfilamente durch Aktin oder Mikrotubuli durch Tubulin). Auch Interaktionen von Proteinen untereinander sind beobachtbar, wenn diese mit verschiedenen fluoreszierenden Proteinen fusioniert werden. Fluoreszierende Proteine können auch unter Kontrolle eines Zelltyp-spezifischen Promotors exprimiert werden, um bestimmte Zelltypen zu identifizieren.[16]
Eine weitere Gruppe bilden Sonden, deren Fluoreszenzverhalten sich in Abhängigkeit vom Zustand ihrer Umgebung ändert. Calcium-abhängige Farbstoffe wie Aequorin, Fura-2, Furaptra, Calcein und Indo-1 können Schwankungen der Konzentration an Calciumionen in einer Zelle anzeigen. Mit spannungsabhängigen Farbstoffen oder den Reporterproteinen VSFP oder PROPS können Spannungsänderungen in einer Zelle dargestellt werden. Mit Redox-sensitiven Reporterproteinen wie roGFP, rxYFP oder HyPer können Redox-Potentiale verfolgt werden. Fluoreszierende pH-Indikatoren können unterschiedliche pH-Werte in einer Zelle sichtbar machen.[17]
- Fluoreszenzmikroskopische Aufnahmen
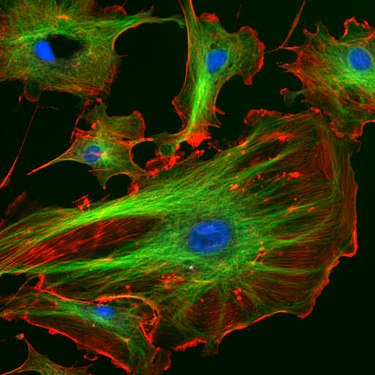 Endothelzellen unter dem Fluoreszenzmikroskop. Die Mikrotubuli sind grün (mit FITC-markiertem Antikörper), Aktinfilamente sind rot (mit Phalloidin-TRITC) markiert worden. Die DNA in den Zellkernen wurde mit DAPI angefärbt (blau).
Endothelzellen unter dem Fluoreszenzmikroskop. Die Mikrotubuli sind grün (mit FITC-markiertem Antikörper), Aktinfilamente sind rot (mit Phalloidin-TRITC) markiert worden. Die DNA in den Zellkernen wurde mit DAPI angefärbt (blau).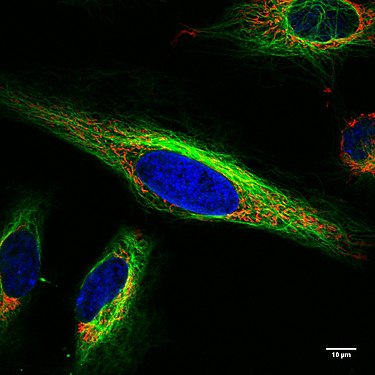 Lebende HeLa-Zellen im Konfokalmikroskop. Mitochondrien in Rot, der Zellkern in Blau, Mikrotubuli in Grün.
Lebende HeLa-Zellen im Konfokalmikroskop. Mitochondrien in Rot, der Zellkern in Blau, Mikrotubuli in Grün.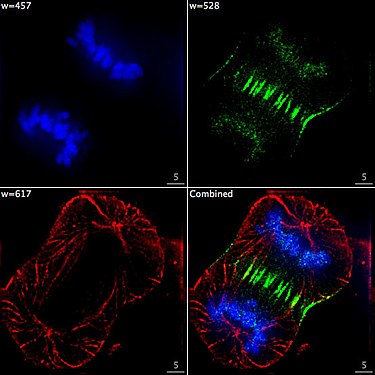 Zelle am Ende der Mitose in der Telophase. Chromosomen blau (DAPI), Mikrotubuli rot, das Protein INCENP ist durch eine Fusion mit GFP grün gefärbt.
Zelle am Ende der Mitose in der Telophase. Chromosomen blau (DAPI), Mikrotubuli rot, das Protein INCENP ist durch eine Fusion mit GFP grün gefärbt. Immunfluoreszenz-Aufnahme im Spinalganglion der Ratte. Zwei verschiedene Proteine wurden mit rot oder grün fluoreszierenden Markern gefärbt.
Immunfluoreszenz-Aufnahme im Spinalganglion der Ratte. Zwei verschiedene Proteine wurden mit rot oder grün fluoreszierenden Markern gefärbt. Metaphasechromosomen aus einem weiblichen menschlichen Lymphozyten, gefärbt mit Chromomycin A3.
Metaphasechromosomen aus einem weiblichen menschlichen Lymphozyten, gefärbt mit Chromomycin A3.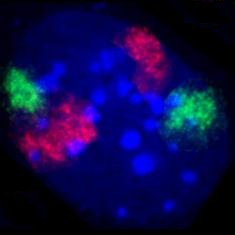 Zellkern eines Mausfibroblasten, durch Fluoreszenz-in-situ-Hybridisierung wurden die Territorien der Chromosomen 2 (rot) und 9 (grün) angefärbt. DNA-Gegenfärbung in blau.
Zellkern eines Mausfibroblasten, durch Fluoreszenz-in-situ-Hybridisierung wurden die Territorien der Chromosomen 2 (rot) und 9 (grün) angefärbt. DNA-Gegenfärbung in blau.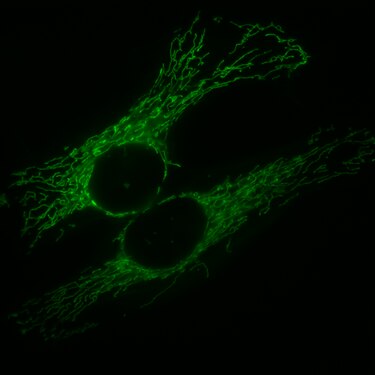 HeLa-Zellen, die Mitochondrien sind mit GFP markiert, welches mit einem entsprechenden Signalpeptid fusioniert ist.
HeLa-Zellen, die Mitochondrien sind mit GFP markiert, welches mit einem entsprechenden Signalpeptid fusioniert ist. Hefezellen, bei denen zwei verschiedene Membranproteine mit GFP oder RFP (Rotes fluoreszierendes Protein) fusioniert sind. Einzelne Zellen haben unterschiedliche Mengen der beiden Proteine und dadurch unterschiedliche Schattierungen.
Hefezellen, bei denen zwei verschiedene Membranproteine mit GFP oder RFP (Rotes fluoreszierendes Protein) fusioniert sind. Einzelne Zellen haben unterschiedliche Mengen der beiden Proteine und dadurch unterschiedliche Schattierungen.

Anwendungen in den Materialwissenschaften
Im Gegensatz zu den Lebenswissenschaften werden in den Materialwissenschaften selten Fluoreszenzfarbstoffe eingesetzt. Die Anwendung von Fluoreszenzmikroskopie beschränkt sich meist auf Fälle, in denen das Material autofluoreszent ist. So sind manche Bestandteile von Verbundwerkstoffen fluoreszierend und können von anderen nicht fluoreszierenden Bestandteilen unterschieden werden. Zur besseren Darstellung der Gegebenheiten kann in einem Konfokalmikroskop Fluoreszenz mit Reflexion kombiniert werden. Dadurch lassen sich etwa in glasfaser-verstärkten Verbundwerkstoffen fluoreszierende Bindemittel (Grundpolymer) von nicht fluoreszierenden Fasern unterscheiden.[18]
Organische Fasern in Papier, Holz oder Bast wurden in etlichen Studien untersucht, um deren Anordnung oder Zusammensetzung zu bestimmen.[19]
Auch verschiedene Typen von Solarzellen können fluoreszenzmikroskopisch untersucht werden. Häufig kommt dabei konfokale Fluoreszenzmikroskopie zum Einsatz, auch in Kombination mit konfokaler Reflexions-Mikroskopie. In organischen Solarzellen kann der Verlust von fluoreszierenden organischen Komponenten untersucht werden, der durch Oxidation mit in die Zelle eingedrungenem Sauerstoff verursacht wird; vorausgesetzt die Abbauprodukte sind nicht fluoreszierend. In Perowskit-Solarzellen wurde analysiert, welchen Einfluss Licht auf die Bildung der gleichnamigen Perowskit-Schicht hat.[20][21][22]

Obwohl etliche Mineralien fluoreszieren, wird Fluoreszenzmikroskopie in der Petrologie wenig genutzt. Eine bemerkenswerte Ausnahme sind Untersuchungen von Kohle und von organischen Einschlüssen in Sedimenten. Reine Mineralien sind meist nicht fluoreszierend, eine Fluoreszenz kann aber durch verschiedene anorganische oder organische Verunreinigungen hervorgerufen werden. Solche gesteinsbildenden Komponenten von Kohle, die einen organischen Ursprung haben, werden als Mazerale bezeichnet. Eine Untergruppe sind die lipidreichen Liptinite. Diese fluoreszierenden Bestandteile können besonders gut an polierten Oberflächen mikroskopisch von nicht-fluoreszierenden anorganischen Bestandteilen unterschieden werden. Während der Kohleentstehung ändern sich die fluoreszenten Eigenschaften, so dass sich Materialien aus verschiedenen Lagerstätten unterscheiden lassen.[23]
Spezielle fluoreszenzmikroskopische Verfahren
Lichtscheibenmikroskopie

Bei der Lichtscheibenmikroskopie (englisch light sheet microscopy; auch Lichtblattmikroskopie oder single plane illumination microscopy, SPIM) wird das Anregungslicht von der Seite als Lichtscheibe beziehungsweise Lichtblatt eingestrahlt. Dies kann durch ein zweites Objektiv oder eine entsprechende Zylinderlinse geschehen, das oder die senkrecht zum Beobachtungsobjektiv steht und eine eng begrenzte Ebene des Präparats ausleuchtet. Nur in dieser ausgeleuchteten Scheibe wird Fluoreszenz erzeugt und diese Ebene wird im Beobachtungsobjektiv scharf gestellt. In anderen Ebenen wird also keine unscharfe Hintergrundfluoreszenz erzeugt, die bei normaler Fluoreszenzmikroskopie zu einer Verminderung des Kontrasts führt. Das Verfahren erlaubt es, die axiale Auflösung eines normalen Fluoreszenzmikroskops zu verbessern, wenn das Lichtblatt dünner als die Schärfentiefe des Beobachtungsobjektivs ist. Die seitliche Beleuchtung des Untersuchungsobjekts ähnelt der Anordnung im Spaltultramikroskop.[24]
Konfokale und Zwei-Photonen-Fluoreszenzmikroskopie

Bei konfokalen wie auch bei Zwei-Photonen-Mikroskopen wird das Präparat abgerastert: Das Anregungslicht wird auf einen Punkt fokussiert, die Fluoreszenz von diesem Punkt gelangt zum Detektor. Der Punkt wird über das Präparat bewegt und die jeweils gemessenen Fluoreszenzintensitäten werden in einem Steuerungscomputer zu einem Bild zusammengesetzt.
Bei der konfokalen Fluoreszenzmikroskopie entsteht im Beleuchtungskegel über und unter der Schärfeebene ebenfalls Fluoreszenz. Diese gelangt jedoch nicht zum Detektor, da sie von einer Lochblende (engl. pinhole) in der Zwischenbild-Ebene blockiert wird. Durch das Blockieren dieser Hintergrundfluoreszenz tritt das Signal in der Schärfeebene gegenüber dem Hintergrund besser hervor als in der klassischen Fluoreszenzmikroskopie. Auf Grund dieser deutlich kontrastreicheren Bilder sind konfokale Mikroskope in der biologischen Forschung weit verbreitet.[25]
Ein Fluoreszenzfarbstoff kann statt durch Absorption eines Photons auch durch eine Zwei-Photonen-Absorption angeregt werden. Voraussetzung dafür ist, dass diese beiden Photonen quasi gleichzeitig am Fluoreszenzfarbstoff eintreffen, und dass beide zusammen den richtigen Energiegehalt haben, um ein Elektron des Farbstoffes auf ein angeregtes Energieniveau zu heben. Beide Bedingungen können erfüllt werden, wenn zur Anregung ein gepulster Laser verwendet wird, der, je nach anzuregendem Fluoreszenzfarbstoff, Wellenlängen zwischen 700 und 1200 nm mit hoher Intensität erzeugt, und dieses Licht durch das Objektiv auf einen Punkt fokussiert wird. Die Photonendichte ist dann so hoch, dass eine ausreichende Wahrscheinlichkeit, dass zwei Photonen gleichzeitig am fluoreszierenden Molekül eintreffen, gegeben ist. Im Gegensatz zur Konfokalmikroskopie, wo das Auslesevolumen beschränkt wird, ist hier das Anregungsvolumen limitiert. Längerwelliges Licht hat eine höhere Eindringtiefe in biologische Gewebe, da es von diesen weniger stark absorbiert und auch weniger stark gestreut wird. Zwei-Photonen-Fluoreszenz-Mikroskopie wird daher eingesetzt, um tiefer in Gewebe einzudringen, als dies mit anderen Verfahren möglich ist. Zusammen mit Methoden, die nicht auf Fluoreszenz beruhen, wird dieses Verfahren als Multiphotonenmikroskopie oder Nicht-lineare Mikroskopie bezeichnet.[26]
Fluoreszenzkorrelationsspektroskopie

Die Fluoreszenzkorrelationsspektroskopie (abgekürzt FCS nach englisch fluorescence correlation spectroscopy) ist zwar eine fluoreszenzmikroskopische Methode, bei ihr wird jedoch kein Bild erzeugt. In einem Konfokalmikroskop wird der Anregungslaser nicht über das Präparat gerastert, sondern auf einer Stelle geparkt. Es wird somit ein sehr kleines Volumen über einen längeren Zeitraum beobachtet. Bewegen sich fluoreszierende Moleküle in dieses Volumen hinein oder hinaus, so ändert sich die gemessene Helligkeit. Anhand einer solchen Messreihe kann beispielsweise bestimmt werden, wie schnell Moleküle in einer Lösung diffundieren. Da die Diffusionsgeschwindigkeit unter anderem von der Größe abhängt, lässt sich beispielsweise untersuchen, ob ein fluoreszenzmarkiertes Protein an ein zweites, ebenfalls in der Lösung vorhandenes Protein bindet und sich dadurch langsamer bewegt.[27]
Verfahren mit erhöhter Auflösung
Als Auflösung wird in der Mikroskopie der Abstand bezeichnet, den zwei Strukturen haben müssen, um als getrennte Strukturen wahrgenommen zu werden. Ernst Abbe hat im 19. Jahrhundert als erster verstanden, dass diese Auflösung fundamental durch Beugung begrenzt ist. Diese Grenze wird daher als Abbe-Limit bezeichnet. Sie kann mit entsprechenden Formeln genau berechnet werden und liegt für gute Mikroskope bei Verwendung von Ölimmersion und in Abhängigkeit von der Wellenlänge bei etwa 200 nm.
Das Abbe-Limit galt lange als unüberwindbar. Ab der zweiten Hälfte des 20. Jahrhunderts wurden jedoch einige mikroskopische Verfahren entwickelt, deren Auflösung besser ist als vom Abbe-Limit vorhergesagt. Das mit Abstand älteste dieser Verfahren ist die Konfokalmikroskopie (siehe auch den Abschnitt Auflösung im Artikel Konfokalmikroskop.) Die theoretische Verbesserung liegt jedoch nur beim Faktor Wurzel von 2 ≈ 1,41. Aus praktischen Gründen kann auch dieser nicht erreicht werden.

In den 1980er-Jahren wurde die TIRF-Mikroskopie (englisch total internal reflection fluorescence microscopy) vorgeschlagen. Mit ihr werden ausschließlich Fluoreszenzfarbstoffe im Präparat angeregt, die sich sehr nah am Deckglas befinden. Ist das Präparat, zum Beispiel lebende Zellen, in wässrigem Medium, so dringt die Anregung vom Deckglas ausgehend nur in eine Schicht von 100–200 nm Dicke ein. Die Schichtdicke ist damit deutlich geringer als es beugungsbedingt mit normaler Mikroskopie möglich wäre. Dadurch ergibt sich ein deutlich höherer Kontrast, da nur wenig Material zur Fluoreszenz angeregt wird. Diese spezielle Form der Anregung wird erreicht, indem das Präparat in einem Winkel angestrahlt wird, der so groß ist, dass an der Kante vom Deckglas zum wässrigen Medium Totalreflexion auftritt und der Lichtstrahl somit gar nicht in das Präparat eindringt. An der Kante tritt jedoch eine evaneszente Welle auf, die zur Anregung führen kann, die sich aber mit zunehmender Entfernung vom Deckglas sehr schnell abschwächt.[24]
Während „Auflösung“ per Definition den Mindestabstand zwischen zwei Strukturen bezeichnet, kann man die genaue Position eines Objekts sehr genau messen. Fluoreszenzmikroskopisch kann man daher das Abbe-Limit umgehen, wenn man die genaue Position von Objekten in verschiedenen Farbkanälen bestimmt und danach den Abstand zwischen diesen misst. Diese Technik wurde in den 1990er-Jahren entwickelt und später als Spektrale Präzisions-Distanz-Mikroskopie (SPDM) bezeichnet. Zwar ließen sich mit ihr Abstände von kleinen fluoreszierenden Strukturen bis auf etwa 70 nm genau messen. Sie führt jedoch nicht zu einer generellen Auflösungsverbesserung, da jeder der aufgenommenen Farbkanäle für sich der Beugung unterliegt.[28][29]
Ende des 20. und Anfang des 21. Jahrhunderts sind jedoch einige Methoden entwickelt worden, mit denen eine generelle Verbesserung möglich ist. Sie werden gemeinschaftlich im Englischen als superresolution microscopy, manchmal auch als nanoscopy bezeichnet. Allen ist gemeinsam, dass sie auf Fluoreszenzmikroskopie beruhen. Drei dieser Verfahren haben eine gewisse Verbreitung erfahren und sind kommerziell erhältlich: STED, strukturierte Beleuchtung und Lokalisationsmikroskopie. Daneben gibt es weitere Verfahren, die sich nicht gegen die genannten behaupten konnten, oder die bisher nur von einzelnen Gruppen angewendet werden. Einige Verfahren werden auf Grund von gemeinsamen Eigenschaften als RESOLFT-Mikroskopie zusammengefasst.
Die hochauflösende Fluoreszenzmikroskopie wurde von der Zeitschrift Nature Methods zur Methode des Jahres 2008 gekürt.[30] William Moerner, Eric Betzig und Stefan Hell erhielten 2014 den Nobelpreis für Chemie für die Entwicklung einiger dieser Methoden.
STED-Mikroskopie

Bei der STED-Mikroskopie (englisch stimulated emission depletion) wird die Beugungsgrenze deutlich überwunden. Der Anregung eines beugungsbegrenzten Volumens im Präparat folgt eine ringförmige Abregung durch Licht längerer Wellenlänge. Dabei fallen die angeregten Moleküle im Bereich der Abregung über stimulierte Emission wieder in den Grundzustand. Das Fluoreszenz emittierende Volumen verkleinert sich dadurch wesentlich und die Auflösung des Mikroskops erhöht sich.[31][24]
Strukturierte Beleuchtung

Bei der strukturierten Beleuchtung oder 3D-SIM-Mikroskopie (englisch structured illumination microscopy) werden nicht alle Fluorochrome angeregt, sondern nur ein Teil des Präparats in Form einer bestimmten ‚Struktur‘, einem Streifenmuster. Bei der Überlagerung des bekannten Beleuchtungsmusters mit der unbekannten Fluorochrom-Verteilung im Präparat entstehen Moiré-Effekte, deren Größe über der Auflösungsgrenze liegt, selbst wenn die unbekannte Struktur kleiner ist. Durch Verschiebung und Verdrehung des Beleuchtungsmusters lässt sich durch die zusätzliche Information aus den Moiré-Mustern der jeweiligen Bilder durch Computerberechnung ein endgültiges Bild mit bis zu zweifach erhöhter Auflösung erzeugen.[32]
Lokalisationsmikroskopie
Als Lokalisationsmikroskopie (englisch localization microscopy) werden mikroskopische Verfahren zusammengefasst, die auf einem gemeinsamen Grundprinzip beruhen: Während bei klassischer Fluoreszenzmikroskopie alle Fluorochrome gleichzeitig angeregt werden, werden sie hier zeitlich nacheinander angeregt, so dass immer nur ein kleiner Teil von ihnen leuchtet. Von einer Schärfeebene werden viele Bilder hintereinander gemacht, oft über Tausend. In jedem dieser Bilder wird nun die genaue Position der jeweils leuchtenden Fluorochrome bestimmt und diese Position wird in das endgültige Bild übertragen. Die Verfahren unterscheiden sich in der Methode, wie die einzelnen Farbstoffmoleküle ein- und ausgeschaltet werden, also zum „Blinken“ gebracht werden.
Die Photoactivated Localization Microscopy (PALM) beruht auf Varianten des Grün Fluoreszierenden Proteins, die sich mit Licht bestimmter Wellenlängen ein- und ausschalten lassen. STORM und dSTORM verwenden geeignete Fluoreszenzfarbstoffe, die in bestimmten Pufferlösungen nur selten fluoreszieren. Ground State Depletion (GSD) beruht darauf, dass sich zu jedem Zeitpunkt die Mehrzahl der Fluorochrome in einem nicht fluoreszierenden Triplett-Zustand befindet, der durch starke Lichtanregung erzeugt werden kann. DNA-Paint beruht auf einer vorübergehenden Bindung von kurzen, einsträngigen DNA-Molekülen an komplementäre Zielmoleküle.[24]
Weitere Verfahren zur Auflösungsverbesserung
4Pi-Mikroskopie war die erste kommerziell verfügbare Superresolution-Technik, ist jedoch heute nicht mehr verfügbar.[24] Eine weitere Technik ist Vertico-SMI, die in Heidelberg entwickelt wurde.
Fluoreszenzlebensdauer-Mikroskopie (FLIM)
Nach der Anregung verbleibt ein fluoreszierender Stoff eine kurze Zeitspanne im angeregten Zustand, bevor er das Fluoreszenzlicht abstrahlt. Die Dauer dieser Zeitspanne variiert für einen konkreten Stoff in einem bestimmten Bereich, so dass eine mittlere Fluoreszenzlebensdauer für einen jeweiligen Stoff bestimmt werden kann. Sie liegt im Nanosekundenbereich, beispielsweise für Fluorescein bei 3,25 ns, für Texas Red bei 3,41 ns und für Eosin bei 1,1 ns. Wenn die Fluoreszenzanregung im mikroskopischen Präparat mit einem gepulsten oder modulierten Laser erfolgt und spezielle Detektoren verwendet werden, dann lässt sich mittels spezieller Messverfahren die Zeitspanne bestimmen, nach welcher die Fluoreszenz am Detektor eintrifft. Nicht nur durch ihre Farbe, sondern auch durch ihre Lebenszeit können daher Fluoreszenzfarbstoffe voneinander unterschieden werden. Dies wird in der Fluoreszenzlebensdauer-Mikroskopie (englisch fluorescence lifetime imaging microscopy, FLIM) ausgenutzt. Gepulste und modulierte Laser gibt es im sichtbaren Wellenlängenbereich für die Anregung mit einem Photon. FLIM kann aber auch in Kombination mit Multi-Photonen-Anregung (siehe oben) verwendet werden, für die ohnehin gepulste Laser erforderlich sind.[33][34]
Förster-Resonanzenergietransfer (FRET)

Beim Förster-Resonanzenergietransfer (manchmal auch: Fluoreszenz-Resonanzenergietransfer) wird die Energie eines angeregten Fluoreszenzfarbstoffs, des Donors, nicht durch Fluoreszenz abgegeben, sondern direkt auf einen anderen Fluoreszenzfarbstoffs (Akzeptor) übertragen. Dies ist möglich, wenn erstens Donor und Akzeptor weniger als 10 nm voneinander entfernt sind und zweitens die Emissionsenergie des Donors der Anregungsenergie des Akzeptors entspricht. Das Emissionsspektrum des Donors muss also mit dem Anregungsspektrum des Akzeptors überlappen. Beispielsweise kann ein grün fluoreszierender Farbstoff als Donor für einen orange fluoreszierenden Akzeptor dienen. Ein weiteres Beispiel ist das cyan fluoreszierende Protein CFP als Donor für das gelb fluoreszierende Protein YFP.[35]
Tritt FRET auf, so wird trotz Anregung des Donors von diesem keine Fluoreszenz ausgesendet, stattdessen kann die Fluoreszenz des Akzeptors beobachtet werden. Die FRET-Effizienz nimmt mit der sechsten Potenz des Abstands zwischen Donor und Akzeptor ab. Das Auftreten von FRET zeigt daher die direkte Nachbarschaft der beiden an, mit einer Genauigkeit, die weit unter der Auflösungsgrenze liegt.[35]
Absichtliches Bleichen zur Diffusionsmessung (FRAP und FLIP)
Bei FRAP (engl. fluorescence recovery after photobleaching) wird ein fluoreszenzmarkiertes Molekül in einer lebenden Zelle in einem Teilbereich der Zelle durch kurzfristige, starke Lichteinwirkung absichtlich gebleicht, meist durch einen fokussierten Laserstrahl. Anschließend wird beobachtet, wie schnell Moleküle aus dem nicht gebleichten Teil der Zelle in den gebleichten Teil zurückkehren. Über die ermittelte Diffusionsgeschwindigkeit können Rückschlüsse auf das Bindungsverhalten des Moleküls gezogen werden.[36]
Bei FLIP (engl. fluorescence loss in photobleaching) wird dagegen ein Bereich der Zelle kontinuierlich gebleicht. Es wird beobachtet, wie schnell die Fluoreszenz im nicht gebleichten Teil der Zelle abnimmt.[36]
Geschichte
1904: Köhlers Entdeckung
Die ersten fluoreszenzmikroskopischen Beobachtungen waren zufällig und ein Ärgernis. August Köhler, Mitarbeiter beim Mikroskophersteller Carl Zeiss, wollte die lichtmikroskopische Auflösung steigern. Die Auflösung hängt von der Wellenlänge ab, daher baute er ein Mikroskop für UV-Licht, um dessen kürzere Wellenlänge zu nutzen. Das damit erzeugte Bild war zwar für die Augen nicht sichtbar, konnte aber fotografisch aufgefangen werden. Er stellte dabei fest, dass
„das Objekt selbst unter dem Einfluß der es treffenden ultravioletten Strahlung fluoresziert. Eine derartige Fluoreszenz tritt aber bei der Mehrzahl der Präparate auf. … Das Fluoreszenzlicht … liegt jedenfalls zum Teil im sichtbaren Bereich des Spektrums;… Daß dadurch die Wahrnehmung des durch die ultravioletten Strahlen erzeugten Bildes, auf die es eigentlich ankommt, erschwert, wenn nicht gar unmöglich gemacht wird, liegt auf der Hand. … Unter Umständen könnte selbstverständlich auch die Beobachtung dieses direkten Bildes Interesse bieten …“
Bereits früh war Köhler also bewusst, dass die bei der UV-Mikroskopie störende Fluoreszenz auch nützliche Anwendungen ermöglichen könnte.
1910–1913: Erste Fluoreszenzmikroskope und Anwendungen

Das erste kommerziell erhältliche Fluoreszenzmikroskop wurde von Oskar Heimstädt entwickelt, der beim Wiener Mikroskopbauer Karl Reichert das Rechen- und Konstruktionsbüro leitete. Es wurde von Reichert persönlich 1911 auf der Versammlung der Deutschen Naturforscher und Ärzte vorgestellt. Im gleichen Jahr veröffentlichte Heimstädt eine Arbeit mit dem Titel „Das Fluoreszenzmikroskop“ in der Zeitschrift für wissenschaftliche Mikroskopie.[37][38]
UV-Licht war für die Fluoreszenzanregung gut geeignet, da es für das Auge nicht sichtbar und somit kein Sperrfilter erforderlich war. Eine gute Anregung erforderte, dass von der Lichtquelle kein sichtbares, aber möglichst viel UV-Licht zum Präparat gelangte. Ein geeigneter Anregungsfilter wurde kurz zuvor, 1910, von Hans Lehmann bei Zeiss entwickelt. Er bestand aus einer Filterküvette, die entlang der optischen Achse aus zwei Kammern bestand. Die drei Wände waren aus Jenaer Blau-Uviolglas, eine Kammer wurde mit gesättigter Kupfersulfat-Lösung, die andere mit verdünnter Nitrosodimethylanilin-Lösung gefüllt. Das so erzeugte Anregungslicht wurde als „gefiltertes Ultraviolett“ bezeichnet.[39] Da normales Glas UV-Licht absorbiert, wurde wenig später ein Kondensor aus Quarz eingesetzt, um hohe Beleuchtungsstärken zu erhalten. Es entstand jedoch ein neues Problem: Die Glaslinsen im Objektiv fingen an zu fluoreszieren. Daher setzte Lehmann einen Dunkelfeldkondensor ein: So wurde das Anregungslicht am Objektiv vorbei geleitet und auch Reste von sichtbarem Licht aus der Lichtquelle im mikroskopischen Bild wurden vermieden.[37][38]
Über die Möglichkeiten und Zukunftsaussichten der Fluoreszenzmikroskopie schrieb Heimstädt
„Es ist z. B. ein leichtes, die Anwesenheit sehr kleiner Mengen von Mutterkorn im Mehl … festzustellen, da Stärke intensiv violett fluoresziert, während Mutterkorn ein gelblich weißes Licht aussendet. …
Ob und wie weit das Fluoreszenzmikroskop … eine Möglichkeit der Erweiterung des mikroskopischen Abbildungsgebietes in sich [schließt], muss die Zukunft lehren.“
Ebenfalls 1911 wurde von Michail Tswett die Fluoreszenz der Chloroplasten beschrieben.[40] Stanislaus von Prowazek veröffentlichte 1913 die erste Arbeit, in der mit Fluoreszenzfarbstoffen gearbeitet wurde, namentlich mit Eosin und Neutralrot.[41]
Ein Jahr nach dem Reichert’schen Fluoreszenzmikroskop wurde auf der Versammlung deutscher Naturforscher und Ärzte 1912 das von Lehmann entwickelte Zeiss’sche „Lumineszenzmikroskop“ vorgestellt. Eine Dunkelfeld-Beleuchtung führt zu einer verringerten Numerischen Apertur und somit zu einer verringerten Auflösung. Daher setzte Lehmann auf eine normale Hellfeldbeleuchtung mit UV-Licht. Um zu verhindern, dass UV-Licht ins Auge drang, setzte er auf einen UV-blockierenden Glasfilter aus Euphosglas, der als Deckglas verwendet wurde. Der Filter musste zwischen Präparat und Objektiv liegen, um Fluoreszenzanregung im Objektiv zu vermeiden, daher war keine andere Position für den Filter möglich. Der Kondensor hatte Quarz-Linsen, die verwendeten Objektträger waren aus Bergkristall, um UV-Durchlässigkeit zu gewährleisten. Die empfohlene Standardvergrößerung lag bei 62× (inklusive Okularvergrößerung), 300× sollte nicht überschritten werden. Die Fluoreszenz-Zusatzausstattung, also ohne das eigentliche Mikroskop, kostete mit der billigsten Anregungslichtquelle, einer von Hand regulierbaren Bogenlampe, etwa 500 Mark. Hinzu kamen 4,50 M für einen 0,5 mm dicken 30 × 25 mm Bergkristall-Objektträger und eine Mark pro Euphosglas-Deckglas. Neben Schwarzweiß-Fotografien konnten auch Farbbilder mit dem Autochromverfahren erstellt werden.[37]
1933–1940: Max Haitinger und die Fluorochromierung

In der Anfangszeit der Fluoreszenzmikroskopie wurden fast ausschließlich autofluoreszente Objekte betrachtet. Auch Max Haitinger, zunächst Privatforscher in Weidling bei Wien, später an der Universität Wien, begann mit der Untersuchung der Fluoreszenz von Wein und Obstweinen. Ab 1933 entwickelte er jedoch Fluoreszenzfärbungen, die er als „Fluorochromierungen“ bezeichnete. Von ihm stammen die Begriffe Sekundärfluoreszenz für zu erzeugende Signale und Primärfluoreszenz für im Präparat von selbst vorhandene. Auch die Bezeichnung Fluorochrom für einen Fluoreszenzfarbstoff wurde von ihm eingeführt. Als Fluorochrome setzte er erst Pflanzenextrakte ein und später eine Reihe von Chemikalien. Dadurch gelang es ihm, Dünnschnitte von tierischen und menschlichen Geweben anzufärben, so dass sich auch Histologen für die Fluoreszenzmikroskopie zu interessieren begannen. Da nur sehr geringe Konzentrationen der Fluorochrome benötigt wurden, waren auch Lebendfärbungen möglich, die hauptsächlich in der Botanik angewendet wurden. Mit Auramin O gelang die Färbung von Tuberkulose-Bakterien. Auch Mehrfachfärbungen waren möglich. Eine erste Abhandlung veröffentlichte Haitinger 1934, sein Buch „Fluorescenzmikroskopie – Ihre Anwendung in der Histologie und Chemie“ erschien 1938.[37][39][42]
Haitinger arbeitete eng mit der Firma Reichert zusammen, um deren Fluoreszenzmikroskop zu verbessern. Das neue Modell „Kam F“ wurde ab 1931 verkauft. Auch dieses hatte eine Bogenlampe mit Eisenelektroden als Lichtquelle, da diese einen vergleichsweise hohen UV-Anteil zwischen 300 nm und 400 nm hatte. Trotzdem benötigte er bis zu 20 Minuten Belichtungszeit für seine Fotografien. Für schwache Vergrößerungen nennt er Belichtungszeiten mit Kohlebogenlampen von einer bis zehn Minuten. Die Nitrosyldimethylanilin-Lösung in der Anregungsfilter-Küvette konnte durch Nickeloxid-haltige Schwarzglasfilter ersetzt werden. Die Kupfersulfat-Lösung, die den verbleibenden Rotanteil im Anregungslicht filterte, wurde erst Anfang der 1940er-Jahre durch blaue Glasfilter ersetzt. Die Beleuchtung erfolgte durch einen Hellfeldkondensor mit normalem Glas; es hatte sich gezeigt, dass Quarzglas hierfür nicht erforderlich ist. Das verbleibende Anregungslicht wurde am Okular blockiert, indem ein Sperrfilter aus 1 mm dickem gelben Glas aufgesetzt wurde. Falls ein farbloser Sperrfilter benötigt wurde, konnte stattdessen eine Küvette mit einer 5 mm dicken Schicht Natriumnitrit-Lösung verwendet werden. Deckgläser aus Euphosglas waren dank des gelben Sperrfilters nicht mehr nötig, auch zeigte sich, dass normale Objektträger geeignet waren. Dadurch sanken die laufenden Kosten erheblich.[37][42]

In seinem Buch von 1938 beschreibt Haitinger auch Auflicht-Beleuchtungen für die Fluoreszenzmikroskopie von undurchsichtigen Gegenständen (Epikondensor von Zeiss-Jena, Epilum der Optischen Werke C. Reichert-Wien (siehe Schemazeichnung), Ultropak von E. Leitz-Wetzlar und Univertor von E. Busch-Rathenow). Sie sind jedoch nicht mit heutiger Auflichtfluoreszenzanregung mit Hilfe eines dichroitischen Strahlteilers vergleichbar. Als Anwendungsbeispiele nennt er die Untersuchung von Nahrungsmitteln, Drogen und Farbstoffen sowie Erstuntersuchungen von Präparaten, von denen Dünnschliffe hergestellt werden sollen. Als „ganz besonders wertvoll“ bezeichnet er Auflichtbeleuchtungen für Studien am lebenden Tier.[42]
Um 1940 brachte Osram Quecksilberhöchstdrucklampen auf den Markt, für die bald bei Zeiss und unter Mitentwicklung von Haitinger bei Reichert (Lux UV und Lux UW) Beleuchtungseinrichtungen ins Programm genommen wurden. Neben der wesentlich vereinfachten Handhabung und einer größeren Helligkeit gaben diese Lampen nicht nur einzelne Linien ab, sondern ein kontinuierliches Spektrum, mit der sich alle fluoreszierenden Stoffe anregen ließen. Auch produzierten sie keine unangenehmen Dämpfe wie Eisenbogenlampen. Vergleichbare Lampen sind auch heute noch in Verwendung.[37][39][43]
In den 1940er-Jahren begann sich Farbdiafilm zur Dokumentation durchzusetzen. Trotz aller Fortschritte lagen die Belichtungszeiten meist noch im zweistelligen Minutenbereich.[37]
1942–1958: Entwicklung der Immunfluoreszenz und die Fluoreszenz herkömmlicher Farbstoffe
Ein Durchbruch für die Fluoreszenzmikroskopie war die Einführung der Immunfluoreszenz. 1942 veröffentlichten Albert Hewett Coons und Kollegen die Kopplung von Fluorescein-Isocyanat an Antikörper. Mit seiner hellen grünen Fluoreszenz hob sich dieses Fluorochrom besser von der bläulichen Autofluoreszenz vieler Gewebe ab als das zuvor probierte Anthracen-Isocyanat. Die Antikörper waren gegen Pneumokokken gerichtet, die so in Mausgeweben fluoreszenzmikroskopisch nachgewiesen werden konnten.[44] Die Kopplung der Antikörper war jedoch technisch anspruchsvoll und die Konjugate waren instabil.[37]
Siegfried Strugger stellte fest, dass manche bekannten herkömmlichen Farbstoffe auch fluoreszieren, so etwa Neutralrot und Rhodamin B. Auch Acridinorange führte er in die Fluoreszenzmikroskopie ein. In einem Buch beschrieb er 1949 detailliert die Möglichkeiten der Fluoreszenzanregung mit blauem Licht statt mit UV. Strugger nutzte Auflichtbeleuchtung, um den Fluss von Wasser in Pflanzen zu verfolgen.[37]
Eine wesentliche Verbesserung der Immunfluoreszenz gelang 1958 J. L. Riggs und Kollegen, indem sie Isothiocyanate statt Isocyanaten verwendeten. Einer anderen Arbeitsgruppe war zwischenzeitlich ein Zwei-Farben-Nachweis mit Fluorescein-Isocyanat und dem orange fluoreszierenden Rhodamin B-Isocyanat gelungen. Riggs und seinen Mitstreitern gelang nun die Markierung von Antikörpern mit den deutlich stabileren Fluoresceinisothiocyanat (FITC) und Rhodamin B-isothiocyanat.[45][37]
1962–1972: Johan Sebastiaan Ploem und die Einführung der Interferenzfilter
Noch in den 1950er-Jahren wurde Fluoreszenzmikroskopie ausschließlich mit UV, violettem oder blauem Anregungslicht durchgeführt.[46][47] Dies änderte sich erst mit der durch den Niederländer Johan Sebastiaan Ploem vorangetriebenen Einführung von Interferenzfiltern in den 1960er-Jahren. Die ersten, die einen dichroitischen Strahlteiler in der Mikroskopie einsetzten, waren die Russen Brumberg und Krylova im Jahr 1953.[48] Die auf Russisch veröffentlichte Arbeit blieb jedoch im Westen unbekannt und wurde erst später „wiederentdeckt“.
Angetrieben durch die Entwicklung zahlreicher Antikörper für Immunfluoreszenzen entstand ein Bedarf an Fluorochromen unterschiedlicher Farben. Diese ließen sich jedoch durch die herkömmliche Verwendung mit UV-Licht oft nur schlecht anregen. Um 1962 begann Ploem eine Zusammenarbeit mit den Schott-Werken in Mainz zur Entwicklung dichroitischer Strahlteiler, die blaues oder grünes Licht reflektierten. Schott stellte zuvor schon viele der in der Fluoreszenzmikroskopie gebräuchlichen Glasfilter her. Die Firma Leitz lieferte Ploem eine Opak-Auflichtbeleuchtung mit einem halbdurchlässigen, farbunabhängigen Spiegel. Diese wurde an der Universiteit van Amsterdam umgebaut und erhielt einen Schieber mit vier Positionen für dichroitische Strahlteiler zur Anregung mit UV, Violett, Blau und Grün, so dass die jeweilige Anregungswellenlänge bequem ausgewählt werden konnte. Erstmals erfolgte die Anregung damit wie heute üblich durch das Objektiv. Durch die Wahl von schmalbandigen Interferenzfiltern für blaues beziehungsweise grünes Anregungslicht konnten außerdem die in der Immunfluoreszenz häufig verwendeten Farbstoffe FITC (grün fluoreszierend) und Tetramethylrhodamin-Isothiocyanat (TRITC; orange fluoreszierend) nahe an ihrem Absorptionsmaximum angeregt werden, ohne gleichzeitig große Mengen Autofluoreszenz durch überflüssige Anregungswellenlängen auszulösen. Seine Ergebnisse veröffentlichte Ploem in mehreren Arbeiten ab 1965.[37][49]


Im Anschluss entwickelte Leitz den PLOEMOPAK, eine Einrichtung, auf der vier Strahlteiler durch Rotation abwechselnd in den Strahlengang eingeschwenkt werden konnten. Spätere Versionen wurden um Sperrfilter und Anregungsfilter ergänzt bis schließlich um 1972 eine Variante auf den Markt kam, die vier Fluoreszenzfilterwürfel mit je einem Anregungsfilter, Strahlteiler und Sperrfilter enthielt. Die Würfel konnten durch den Anwender ausgetauscht werden, um sie den jeweils verwendeten Fluorochromen anzupassen.[49]
Damit war die Entwicklung des Epifluoreszenzmikroskops, wie es heute noch verwendet wird, im Prinzip abgeschlossen. Es sollte jedoch noch etliche Jahre dauern, bis sich dieser Bautyp allgemein durchsetzte. Britische Mikroskopie-Lehrbücher von 1975 und 1977 erwähnen ausschließlich die Möglichkeit der UV-Anregung und Durchlichtbeleuchtung.[50][51] Ein späteres von 1982 meinte, dass die Beleuchtung für gewöhnlich mit einem Dunkelfeldkondensor erfolge, beschrieb aber auch die Auflichtbeleuchtung mit dichroitischem Strahlteiler und stellte diese als die lichtempfindlichere vor. Die Möglichkeit, durch den Austausch aller Filter Fluorochrome wie FITC und Rhodamin nacheinander nachweisen zu können, wurde ebenfalls erwähnt.[52]
Ein westdeutsches Lehrbuch von 1985 beschrieb alle drei Möglichkeiten, Durchlicht-Hell- und -Dunkelfeld sowie Auflicht-Hellfelderregung durch das Objektiv, schrieb letzterer besonders einfache Einstellung (da kein Kondensor benötigt wird) sowie hohe Anregungsintensität bei ausgezeichnetem Kontrast zu und erwähnte die Möglichkeit, Filterblöcke schnell auszutauschen. Als Hersteller solcher Systeme wurden Leitz, Olympus, Reichert, Zeiss und Jena genannt, womit wohl der VEB Carl Zeiss Jena gemeint war.[53] Ein Lehrbuch von 1988 erwähnte zwar die älteren Methoden, stellte dann aber fest: „Moderne Fluoreszenzmikroskope arbeiten nach dem Prinzip der Auflicht-Hellfeldanregung“ mit dichroitischer Teilerplatte. Im Weiteren hieß es: „Die meisten Mikroskophersteller bieten Einrichtungen für die Durchlicht- und Auflicht-Fluoreszenzmikroskopie an“.[54]
Weblinks
- Datenbanken für Fluoreszenzfarbstoffe: Fluorophores.org, Spectra Database an der University of Arizona, Fluorescence SpectraViewer bei Thermo Fisher.
- Michael Volger (Hrsg.: Irene K. Lichtscheidl): Fluoreszenzmikroskopie. Abgerufen am 25. März 2017 (Theoretische Einführung und Anleitung zur praktischen Anwendung an der Uni Wien. univie.ac.at (PDF; 2,5 MB)).
- Multi-Wavelength Epi-Illumination in Fluorescence Microscopy by Johan Sebastiaan Ploem and Friedrich Walter on Leica Science Lab.
Einzelnachweise
- ↑ a b Jörg Haus: Optische Mikroskopie. Wiley-VCH, Weinheim 2014, ISBN 978-3-527-41127-6, S. 163–173.
- ↑ a b c d e f g h i j k l m Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 35–48.
- ↑ Markus Axmann, Josef Madl, Gerhard J. Schütz: Single-Molecule Microscopy in the Life Sciences. In: Ulrich Kubitscheck (Hrsg.): Fluorescence Microscopy. Wiley-Blackwell, Weinheim 2013, ISBN 978-3-527-32922-9, S. 293–343, hier S. 309.
- ↑ I. Georgakoudi, B. C. Jacobson, M. G. Müller, E. E. Sheets, K. Badizadegan, D. L. Carr-Locke, C. P. Crum, C. W. Boone, R. R. Dasari, J. Van Dam, M. S. Feld: NAD(P)H and collagen as in vivo quantitative fluorescent biomarkers of epithelial precancerous changes. In: Cancer research. Band 62, Nummer 3, Februar 2002, S. 682–687, PMID 11830520.
- ↑ M. R. Speicher, S. Gwyn Ballard, D. C. Ward: Karyotyping human chromosomes by combinatorial multi-fluor FISH. In: Nature genetics. Band 12, Nummer 4, April 1996, S. 368–375, doi:10.1038/ng0496-368. PMID 8630489.
- ↑ A. Bolzer, G. Kreth, I. Solovei, D. Koehler, K. Saracoglu, C. Fauth, S. Müller, R. Eils, C. Cremer, M. R. Speicher, T. Cremer: Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. In: PLoS biology. Band 3, Nummer 5, Mai 2005, S. e157, doi:10.1371/journal.pbio.0030157. PMID 15839726, PMC 1084335 (freier Volltext).
- ↑ a b Douglas B. Murphy, Michael W. Davidson: Fundamentals of Light Microscopy and Electronic Imaging. 2. Auflage. Wiley-Blackwell, Hoboken NJ 2013, ISBN 978-0-471-69214-0, S. 45–50.
- ↑ Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 75–81.
- ↑ Douglas B. Murphy, Michael W. Davidson: Fundamentals of Light Microscopy and Electronic Imaging. 2. Auflage. Wiley-Blackwell, Hoboken NJ 2013, ISBN 978-0-471-69214-0, S. 389–413.
- ↑ Rolf Theodor Borlinghaus: Konfokale Mikroskopie in Weiß: Optische Schnitte in allen Farben. Springer Spektrum, 2016, ISBN 978-3-662-49358-8.
- ↑ Ian D. Johnson: Practical Considerations in the Selection and Application of Fluorescent Probes. In: James Pawley (Hrsg.): Handbook of Biological Confocal Microscopy. 3. Auflage. Springer Science and Business Media LLC, 2006, ISBN 0-387-25921-X, Kapitel 1, S. 353–367.
- ↑ a b Douglas B. Murphy, Michael W. Davidson: Fundamentals of Light Microscopy and Electronic Imaging. 2. Auflage. Wiley-Blackwell, Hoboken NJ 2013, ISBN 978-0-471-69214-0, S. 361–365.
- ↑ a b c Gerd Ulrich Nienhaus, Karin Nienhaus: Fluorescence Labeling. In: Ulrich Kubitscheck (Hrsg.): Fluorescence Microscopy. Wiley-Blackwell, Weinheim 2013, ISBN 978-3-527-32922-9, S. 143–173, hier S. 147–148.
- ↑ Douglas B. Murphy, Michael W. Davidson: Fundamentals of Light Microscopy and Electronic Imaging. 2. Auflage. Wiley-Blackwell, Hoboken NJ 2013, ISBN 978-0-471-69214-0, S. 227 f.
- ↑ a b Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 181–190.
- ↑ Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 169–180.
- ↑ Douglas B. Murphy, Michael W. Davidson: Fundamentals of Light Microscopy and Electronic Imaging. 2. Auflage. Wiley-Blackwell, Hoboken NJ 2013, ISBN 978-0-471-69214-0, S. 167.
- ↑ Ashley R. Clarke, Colin Nigel Eberhardt: Microscopy Techniques for Materials Science. Woodhead Publishing, Abington Hall 2002, ISBN 978-1-85573-587-3, S. 255, 279–284.
- ↑ L.A. Donaldson: Analysis of fibres using microscopy. In: Handbook of Textile Fibre Structure. Fundamentals and Manufactured Polymer Fibres. Volume 1 in Woodhead Publishing Series in Textiles. 2018 Elsevier, 2009, S. 121–153, doi:10.1533/9781845696504.1.121.
- ↑ Pankaj Kumar: Organic Solar Cells: Device Physics, Processing, Degradation, and Prevention. CRC Press, 2016, ISBN 978-1-4987-2327-5, S. 23 (Google Books).
- ↑ J. H. Huang, F. C. Chien, P. Chen, K. C. Ho, C. W. Chu: Monitoring the 3D nanostructures of bulk heterojunction polymer solar cells using confocal lifetime imaging. In: Analytical Chemistry. Band 82, Nummer 5, März 2010, S. 1669–1673, doi:10.1021/ac901992c, PMID 20143827.
- ↑ A. Ummadisingu, L. Steier, J. Y. Seo, T. Matsui, A. Abate, W. Tress, M. Grätzel: The effect of illumination on the formation of metal halide perovskite films. In: Nature. Band 545, Nummer 7653, 05 2017, S. 208–212, doi:10.1038/nature22072, PMID 28445459.
- ↑ F. W. D. Rost: Fluorescence Microscopy Volume II. Cambridge University Press, 1995, ISBN 978-0-521-41088-5, S. 40–47 (online bei Google Books).
- ↑ a b c d e Jörg Haus: Optische Mikroskopie. Wiley-VCH, Weinheim 2014, ISBN 978-3-527-41127-6, S. 189–200.
- ↑ Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 63 ff.
- ↑ Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 109 ff.
- ↑ Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 223 ff.
- ↑ Harald Bornfleth, Kurt Satzler, Roland Eils, Christoph Cremer: High-precision distance measurements and volume-conserving segmentation of objects near and below the resolution limit in three-dimensional confocal fluorescence microscopy. In: Journal of Microscopy. Band 189, Nr. 2, Februar 1998, S. 118, doi:10.1046/j.1365-2818.1998.00276.x.
- ↑ Steffen Dietzel, Roland Eils, Kurt Sätzler, Harald Bornfleth, Anna Jauch, Christoph Cremer, Thomas Cremer: Evidence against a Looped Structure of the Inactive Human X-Chromosome Territory. In: Experimental Cell Research. Band 240, Nr. 2, Mai 1998, S. 187, doi:10.1006/excr.1998.3934, PMID 9596991.
- ↑ Method of the Year 2008. In: Nature Methods, 6, 2009, S. 1, doi:10.1038/nmeth.f.244.
- ↑ Volker Westphal, Silvio O. Rizzoli, Marcel A. Lauterbach, Dirk Kamin, Reinhard Jahn, Stefan W. Hell: Video-Rate Far-Field Optical Nanoscopy Dissects Synaptic Vesicle Movement. In: Science. Band 320, Nr. 5873, 11. April 2008, S. 246–249, doi:10.1126/science.1154228.
- ↑ Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 245–247.
- ↑ Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 207 ff.
- ↑ Douglas B. Murphy, Michael W. Davidson: Fundamentals of Light Microscopy and Electronic Imaging. 2. Auflage. Wiley-Blackwell, Hoboken NJ 2013, ISBN 978-0-471-69214-0, S. 280 ff.
- ↑ a b Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 213 ff.
- ↑ a b Guy Cox: Optical Imaging Techniques in Cell Biology. 2. Auflage. CRC Press, Taylor & Francis Group, Boca Raton FL 2012, ISBN 978-1-4398-4825-8, S. 201 ff.
- ↑ a b c d e f g h i j k l Dieter Gerlach: Geschichte der Mikroskopie. Verlag Harri Deutsch, Frankfurt am Main 2009, ISBN 978-3-8171-1781-9, S. 625–657.
- ↑ a b c Oskar Heimstädt: Das Fluoreszenzmikroskop. In: Zeitschrift für wissenschaftliche Mikroskopie. Band 28, 1911, S. 330–337 (online).
- ↑ a b c Fritz Bräutigam, Alfred Grabner: Fluoreszenzmikroskopie. In: Fritz Bräutigam, Alfred Grabner (Hrsg.): Beiträge zur Fluoreszenzmikroskopie (= 1. Sonderband der Zeitschrift „Mikroskopie“). Verlag Georg Fromme & Co., Wien 1949, S. 25–34.
- ↑ Michail Tswett: Über Reicherts Fluoreszenz-Mikroskop und einigen damit angestellten Beobachtungen über Chlorophyll und Cyanophyll. In: Berichte der Deutschen Botanischen Gesellschaft. Band 29, 1911, S. 744–746. (online – Zitiert nach Gerlach, 2009.)
- ↑ Stanislaus von Prowazek: Fluorescenz der Zellen. – Reicherts Fluoreszenzmikroskop. In: Zoologischer Anzeiger. Band 42, 1913, S. 374–380. (online – Zitiert nach Gerlach, 2009.)
- ↑ a b c d Max Haitinger: Fluorescenzmikroskopie-Ihre Anwendung in der Histologie und Chemie. Akademische Verlagsgesellschaft, Leipzig 1938.
- ↑ Karl Höfler: Max Haitinger 1868–1946. In: Hugo Freund, Alexander Berg (Hrsg.): Angewandte Naturwissenschaften und Technik (= Geschichte der Mikroskopie Leben und Werk großer Forscher. Band III). Umschau Verlag, Frankfurt am Main 1966, S. 187–194.
- ↑ Albert H. Coons, Hugh J. Creech, R. Norman Jones, Ernst Berliner: The Demonstration of Pneumococcal Antigen in Tissues by the Use of Fluorescent Antibody. In: Journal of Immunology. Band 45, Nr. 3, 1. November 1942, S. 159–170 (online).
- ↑ J. L. Riggs, R. J. Seiwald, J. H. Burckhalter, C. M. Downs, T. G. Metcalf: Isothiocyanate compounds as fluorescent labeling agents for immune serum. In: The American Journal of Pathology. Band 34, Nummer 6, Nov-Dez 1958, S. 1081–1097. PMID 13583098, PMC 1934794 (freier Volltext).
- ↑ E. S. Perner: Die Methoden der Fluoreszenzmikroskopie. In: Hugo Freund (Hrsg.): Die optischen Grundlagen, die Instrumente und Nebenapparate für die Mikroskopie in der Technik. Teil 1: Allgemeines Instrumentarium der Durchlichtmikroskopie (= Handbuch der Mikroskopie in der Technik. Band 1, Teil 1). Umschau Verlag, Frankfurt am Main 1957, S. 357–431, hier S. 371.
- ↑ Heinz Appelt: Einführung in die mikroskopischen Untersuchungsmethoden. 4. Auflage. Akademische Verlagsgesellschaft Geest & Portig, Leipzig 1959, S. 283.
- ↑ E. M. Brumberg, T. N. Krylova: O fluoreschentnykh mikroskopopak. In: Zh. obshch. biol. 14, 1953, S. 461.
- ↑ a b Johan Sebastiaan Ploem, Friedrich Walter: Multi-Wavelength Epi-Illumination in Fluorescence Microscopy. In: Leica Microsystems (Hrsg.): Scientific and Technical Information CDR. Band 5, 2001, S. 1–16 (leica-microsystems.com/science-lab).
- ↑ H. N. Southworth: Introduction to Modern Microscopy. Wykeham Publications LTD, a member of the Taylor & Francis Group, London 1975, ISBN 0-85109-470-8, S. 84–85.
- ↑ W. Burrels: Microscope Technique. A Comprehensive Handbook for General and Applied Microscopy. New revised edition Auflage. Fountain Press, London 1977, ISBN 0-85242-511-2, S. 481–483.
- ↑ Michael Spencer: Fundamentals of light microscopy (= IUPAB Biophysics Series. Band 6). Cambridge University Press, Cambridge, England 1982, ISBN 0-521-28967-X, S. 40–45.
- ↑ Dieter Gerlach: Das Lichtmikroskop. Eine Einführung in Funktion und Anwendung in Biologie und Medizin. 2. Auflage. Thieme Verlag, Stuttgart 1985, ISBN 3-13-530302-0, S. 210–224.
- ↑ Gerhard Göke: Moderne Methoden der Lichtmikroskopie: vom Durchlicht-Hellfeld- bis zum Lasermikroskop. Franckh’sche Verlagshandlung, Stuttgart 1988, ISBN 3-440-05765-8, S. 211–212.
Auf dieser Seite verwendete Medien

Endothelzellen aus der Inneren Wand (Endothel) von Lungenarterien des Rindes unter dem Mikroskop. Die Zellkerne sind mit DAPI blau markiert. Die Mikrotubuli wurden über einen Antikörper grün markiert. Mit rot fluoreszierendem Phalloidin wurden die Aktinfilamente markiert.

Autor/Urheber: Krzysztof Blachnicki, derivative work: Henry Mühlpfordt. derivative work: User:Dietzel65, Lizenz: CC BY-SA 3.0
Schema eines Auflichtmikroskops
Autor/Urheber: Dietzel65, Lizenz: CC BY-SA 4.0
Mikroskopische Aufnahme des Mooses Plagiomnium undulatum. Oben: Hellfeld-Aufnahmne. Unten: Autofluoreszenz der Chloroplasten. Einzelbilder, aufgenommen mit einem 40x0.8 Objektiv.

Autor/Urheber: 8x57is, Lizenz: CC BY-SA 4.0
Konfokales Bild (cLSM) lebender Hela Zellen. Mitochondrien sind rot gefärbt (Mitotracker red), der Zellkern ist blau (DAPI).Das Mikrotubulinetzwerk ist in grün gefärbt (Tubulintracker green). Mitochondrien bilden Netzwerke und werden mittlels Motorproteinen entlang Mikrotubuli transportiert.
Autor/Urheber: Jacobkhed, modified by Dietzel65, Lizenz: CC0
Jablonski-Diagramm speziell für Fluoreszenz
Autor/Urheber: Zadelrob, Lizenz: CC BY-SA 3.0
Excitation and emission spectra of 10 µM fluorescein in deionized water.
Autor/Urheber: self, Lizenz: CC BY-SA 4.0
Fluoreszenz-Filterwürfel für grüne Fluorochrome. Filter system L5 ET: BP 480/40; LP 505, BP 527/30. Links unten: Blick durch den Anregungsfilter auf einen bewölkten Himmel. Rechts unten: Selbiges durch den Emissionsfilter

Autor/Urheber: Helmut Hund GmbH, Rechtsnachfolger von Wild, Lizenz: CC BY-SA 4.0
Katalog der Firma Will zu Fluoreszenzmikroksopen (Auflicht, Durchlicht), Seite 1/4
Autor/Urheber: Xibo Wang1, Lili Zhang1, Yaofa Jiang2, Jianpeng Wei1, and Zijuan Chen1, from the following institutions: 1) State Key laboratory of Coal Resources and Safety Mining, China University of Mining and Technology, Beijing 100083, China; 2) Jiangsu Institute of Architectural Technology, Xuzhou 221116, China, Lizenz: CC BY 4.0
Macerals in the Upper No. 3 coals. Left: Sporinite, corpogelinite, inertodetrinite, and colledetrinite (sample no. LBS3U-4), under oil reflectance white light; right: Sporinite under fluorescent light (same sample).
Autor/Urheber: Dietzel65, Steffen Dietzel, Lizenz: CC BY 3.0
Einzelner optischer Schnitt einer fixierten HeLa-Zelle, die einem scratch-replication-label unterzogen wurde, durch das die Orte der DNA-Synthese markiert wurden (rot). Links: konfokale Aufnahme (pinhole = 1 Airy unit). Rechts: nicht-konfokale Aufnahme mit völlig geöffnetem Pinhole.
Autor/Urheber: Biooid 99, Lizenz: CC BY-SA 4.0
Norway spruce seedling hypocoty cross sectionl age 6 weeks, autofluorescence.
Autor/Urheber:
unbekannt
, Lizenz: PD-alt-100{kind=link}
Das erste kommerzielle Fluoreszenzmikroskop. Hergestellt von der Firma Reichert in Wien und auf den Markt gebracht 1911.
Simplyfied Jablonski energy diagram for a Förster resonance energy transfer.
Blue: donor excitation Green: aceptor emission
Black: radiation-freeAutor/Urheber: Dietzel65, Lizenz: CC0
Schieber für ein Fluoreszenzmikroskop mit Filtern für drei Fluoreszenkanäle.
Autor/Urheber: Fabian Göttfert, Christian Wurm, Lizenz: CC BY-SA 3.0
This image shows the resolution gain possible with STED microscopy over confocal microscopy. Confocal raw data is on the left side and STED raw data on the right. The dye is Abberior STAR635P. The optical resolution of the STED image is 25nm or smaller as can be seen from diameter of the single dots (antibody clusters).
Autor/Urheber: Mad scientist, Lizenz: CC BY-SA 3.0
Evaneszente Welle in der TIRF (total internal reflection) Mikroskopie, grobes Funktionsschema. Die Proportionen sind zur besseren Verständlichkeit nicht realistisch. Beschriftung in Deutsch.
Autor/Urheber: Jacobkhed, modified by Dietzel65, Lizenz: CC0
Jablonski-Diagramm speziell für Fluoreszenz, mit S2- und T1-Zustand.

Autor/Urheber: transfert on commons by F Lamiot, Lizenz: CC BY-SA 4.0
Dividing Cell Fluorescence ; An image of a Human cancer cell dividing. Taken with an epifluorescence microscope. The cell is fixed and stained with DAPI (Stains DNA blue), GFP-INCENP (green), Tubulin (red).
Autor/Urheber: Dietzel65, Lizenz: CC BY-SA 4.0
Pollenkorn von Hibiscus. Autofluoreszenz in 3 Farbkanälen, angeregt mit 405 nm. Projektion von konfokalen Aufnahmen.
Autor/Urheber: Dietzel65, Lizenz: CC BY-SA 4.0
Absorptionsspektrum (blau), Emissionsspektrum (rot) und die der jeweilige Elektronenübergang, der den Peak hervorruft bei einem hypothetischen Fluorochrom.

(c) Nmnogueira in der Wikipedia auf Englisch, CC BY-SA 2.5
Inverted microscope arranged for fluorescence microscopy. Nikon TE2000S.

Autor/Urheber: Dietzel65, Lizenz: CC0
Leica DM 2500 Fluoreszenzmikroskop mit abgenommener Abdeckung, so dass der Revolver mit den Fluoreszenzfilterwürfeln zu sehen ist.

Autor/Urheber: Steffen Dietzel, Lizenz: CC BY-SA 3.0
Metaphasechromosomen aus einer weiblichen menschlichen Lymphozytenzelle, gefärbt mit Chromomycin A3, fluoreszenzmikroskopische Aufnahme. Hergestellung: Colcemid-Behandlung, Hypotonie, Methanol-Eisessig Fixierung, Auftropfen, Lufttrocknen, Färbung.
Originallegende: "Wurzelspitze von Allium cepa, längs, mit Coriphosphin O; Reichert-Objektiv 3, Okular I, 10 Minuten Belichtungszeit, scharfe Differenzierung zwischen Kern und Protoplasma, in den Farben wie bei Fig. 12, bei stärkerer Vergrößerung sieht man Kern und Plasmastruktur. Reichert Kam. VY" Anmerkung: gemeint ist in den Farben wie Fig. 11. Dort steht: Plasma orange, Kerne gelb.

Autor/Urheber: Robert Mayer, Alessandro Brero, Johann von Hase, Timm Schroeder, Thomas Cremer, Steffen Dietzel, Lizenz: CC BY 2.0
Zellkern eines Mausfibroblasten, durch Fluoreszenz in situ Hybridisierung wurden die Territorien der Chromosomen 2 (rot) und 9 (grün) angefärbt. DNA-Gegenfärbung in blau.

Autor/Urheber: Masur, Lizenz: CC BY 2.5
Yeast (S. cerevisiae) cell membrane visualized by some membrane proteins fused with RFP and GFP fluorescent markers. Imposition of light from both of markers results in yellow colour.
Autor/Urheber: Andreas Bolzer, Gregor Kreth, Irina Solovei, Daniela Koehler, Kaan Saracoglu, Christine Fauth, Stefan Müller, Roland Eils, Christoph Cremer, Michael R. Speicher, Thomas Cremer, Lizenz: CC BY 2.5
Zellkern eines menschlichen Fibroblasten, in dem alle 24 verschiedenen Chromosomen (1 - 22, X und Y) per Fluoreszenz in situ Hybridisierung (FISH) mit einer unterschiedlichen Kombination von insgesamt 7 Fluorochromen angefärbt wurden. Gezeigt ist eine mittlere Ebene in einem deconvolvierten Bildstapel, der mit Weitfeld-Mikroskopie aufgenommen wurde.

Autor/Urheber: Simon Troeder, Lizenz: CC BY 4.0
Fluorescent microscopy picture of HeLa cells expressing a mitochondrially targeted version of green fluorescent protein (mtGFP)

Autor/Urheber: Masur, Lizenz: CC BY-SA 3.0
An Olympus BX61 (epi-)fluorescence microscop coupled with a digital camera.

Autor/Urheber: Der ursprünglich hochladende Benutzer war Zsynth in der Wikipedia auf Deutsch, Lizenz: CC BY-SA 3.0
Immunfluoreszenz-Aufnahme im Spinalganglion der Ratte. Zwei verschiedene Proteine wurden mit rot oder grün fluoreszierenden Markern gefärbt.
Autor/Urheber: Changes in layout by the uploader. Only the creator of the original (Lothar Schermelleh) should be credited., Lizenz: CC BY-SA 3.0
Vergleich des Auflösungsvermögens von konfokaler Laser-Scanning Mikroskopie (CLSM, oben) und 3D-SIM (unten). Zellkernporen (anti-NPC, rot), Zellkernhülle (anti-Lamin B, grün), sowie DNA verpackt in Chromatin (DAPI, blau) wurden in einer Mauszelle simultan angefärbt. Der Maßstab entspricht 1 µm. )